
بیماری SMA چیست؟
با بیماری آتروفی عضلانی نخاعی (SMA) آشنا شوید!
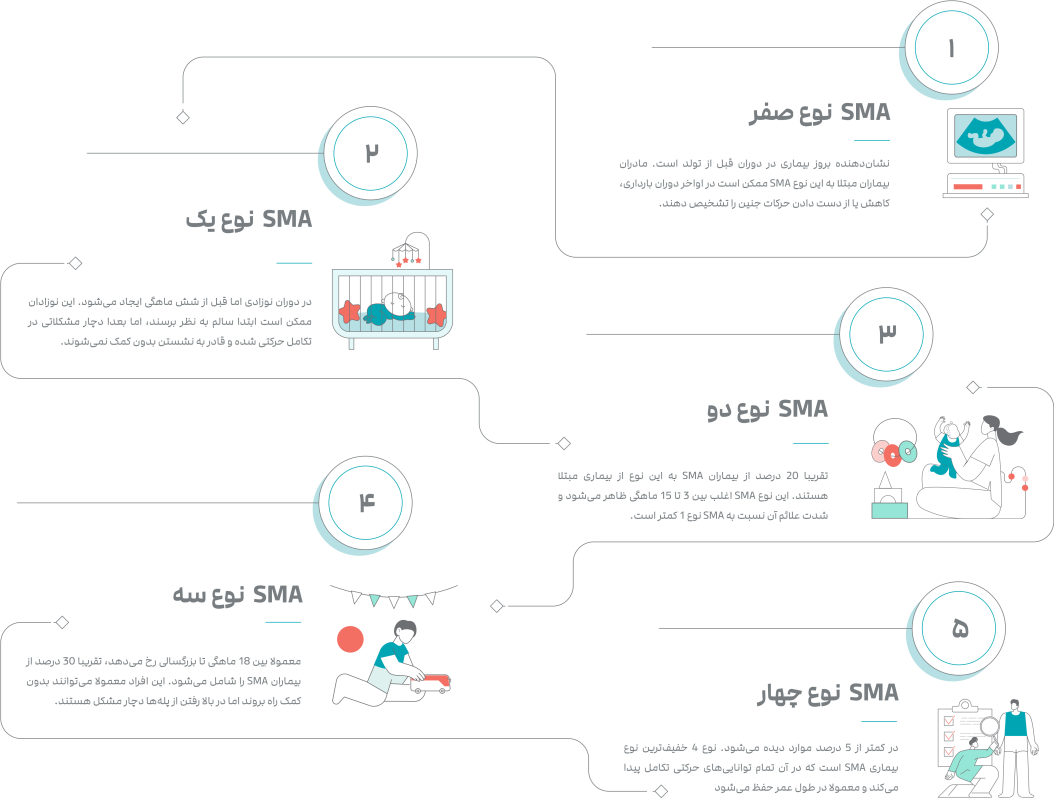
اس ام ای نوع صفر
اس ام ای نوع یک
اس ام ای نوع دو
اس ام ای نوع سه
اس ام ای نوع چهار
علت آتروفی عضلات نخاعی
اپیدمیولوژی SMA
بر اساس مطالعات، فراوانی افراد ناقل با درنظرگیری تنوع جمعیتی، از یک نفر در گروه جمعیتی 100 نفره (1 درصد) تا یک نفر در گروه جمعیتی 45 نفره (کمی بیش از 2 درصد) متغیر است. طبق برآوردها بیماری SMA در هر 100 هزار تولد، 5 تا 13 نفر را تحت تاثیر قرار میدهد. در کشور ایران بیش از 900 بیمار SMA ثبت شدهاند.

مدیریت و درمان SMA
درمان کاملی برای این بیماری وجود ندارد و فقط میتوان سرعت پیشرفت آن را کند کرده و کیفیت زندگی را بهبود بخشید. انتخاب روش درمانی به نوع SMA، شدت بیماری و در دسترس بودن دارو وابسته است.
1) درمانهای اصلاحکننده بیماری
داروهای Risdiplam (Evrysdi®) و Nusinersen (Spinraza®) تولید پروتئین SMN را تحریک میکنند و توسط سازمان غذا و دارو آمریکا برای مصرف کودکان و بزرگسالان تایید شدهاند.
2) ژن درمانی
داروی Onasemnogene abeparvovec (Zolgensma®) ژن SMN1 معیوب و یا از دست رفته را با یک ژن فعال جایگزین میکند.

واحد پشتیبانی از بیمار
شما میتوانید سوالات خود را درباره SMA یا اورلیژن در روناکر از متخصص بپرسید.
سوالات مربوط به بیماری SMA
آتروفی عضلانی نخاعی (Spinal Muscular Atrophy; SMA) یک بیماری ژنتیکی است که به علت از بین رفتن نورونهای حرکتی ایجاد میشود. از دست دادن این سلولهای عصبی در بیماران مبتلا به SMA، منجر به ضعف عضلانی پیشرونده، کاهش حجم عضلات (آتروفی) و کاهش تون عضلانی (هیپوتونی) میشود. به همین دلیل ممکن است مشکلاتی در کنترل حرکات بدن، ایستادن، نشستن و حتی بلع و تنفس پدیدار شود.
نورونهای حرکتی برای عملکرد صحیح خود نیاز به تولید پروتئینی به نام Survival Motor Neuron (SMN) دارند. این پروتئین حاصل فعالیت ژنهایی به نام SMN1 و SMN2 است. در بیماران مبتلا به SMA، به علت حذف بخشی از ژن SMN1 یا جهش ژنی، پروتئین SMN به میزان کافی تولید نمیشود و به همین علت، نورونهای حرکتی کوچک و ضعیف میشوند و در نهایت از بین میروند.
بیماری SMA بر اساس سن بروز و سیر بالینی علائم به انواع 0 تا 4 طبقه بندی میشود.
SMA نوع 0 (بروز بیماری قبل از تولد) و SMA نوع 1 (بروز بیماری در دوران نوزادی) شایعترین و شدیدترین انواع SMA هستند. SMA نوع 2 و SMA نوع 3 بروز دیرتر و شدت علائم کمتری دارند. SMA نوع 4 (بروز بیماری در دوران بزرگسالی) کمترین شدت علائم بیماری را در مقایسه با سایر دستهها دارد.
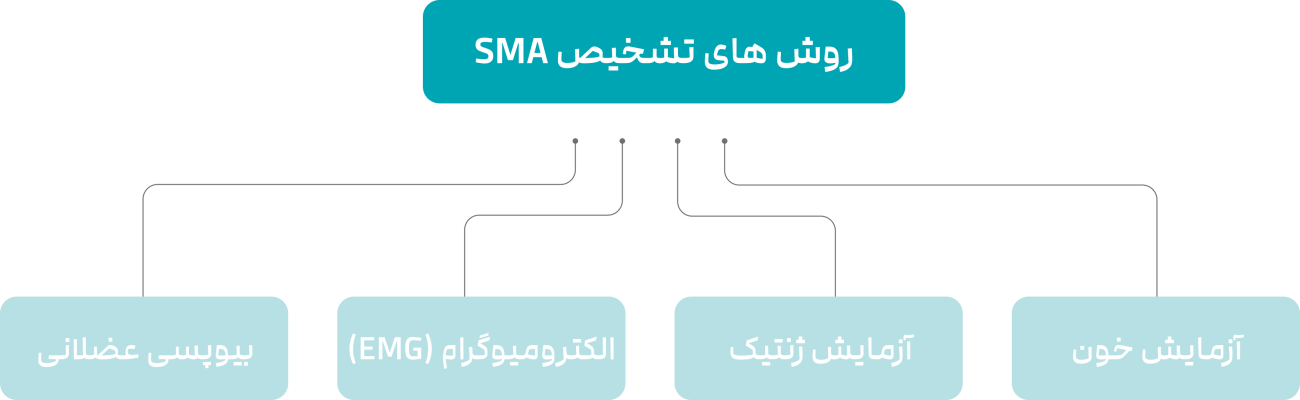
علاوه بر انجام معاینههای فیزیکی، پزشک متخصص ممکن است آزمایش خون، آزمایش ژنتیک، الکترومیوگرام یا نمونهبرداری عضلانی را برای تشخیص بیماری SMA درخواست کند.
با توجه به این که بیماری SMA به صورت ژنتیکی انتقال پیدا میکند، میتوان قبل از بارداری با غربالگری افراد از نظر ژن ناقل SMA، از بروز این بیماری جلوگیری کرد. افرادی که با دو نسخه از ژن معیوب متولد میشوند مبتلا به بیماری خواهند بود.
فراوانی افراد ناقل با درنظرگیری تنوع جمعیتی، از یک نفر در گروه جمعیتی 100 نفره (1 درصد) تا یک نفر در گروه جمعیتی 45 نفره (کمی بیش از 2 درصد) متغیر است. طبق برآوردها بیماری SMA در هر 100 هزار تولد، 5 تا 13 نفر را تحت تاثیر قرار میدهد.
SMA معمولا در مردان و زنان به طور برابر رخ میدهد.
ناقل فردی است که یک نسخه سالم و یک نسخه معیوب از ژن SMN1 را به ارث میبرد. اگر پدر و مادر هر دو ناقل باشند، احتمال داشتن فرزند مبتلا به SMA حدود 25 درصد است.
به طور کلی، ناقلین SMA علائم بیماری را در خود نشان نمیدهند. بیشتر ناقلین تا زمانی که فرزندی مبتلا به SMA نداشته باشند، از داشتن مشکل ژنتیکی خود آگاه نیستند.
بله، SMA یک بیماری ارثی بوده و اگر یکی از اعضای خانواده شما مبتلا به SMA باشد، به این معنی است که خطر ناقل بودن شما و احتمال ابتلای فرزندانتان افزایش پیدا میکند.
غربالگری ناقل نوعی آزمایش است که میتواند شما را از نظر داشتن ژنهای تغییر یافته برای برخی اختلالات ژنتیکی بررسی کند.
غربالگری SMA میتواند به شما برای پیشبینی این که آیا کودک مبتلا به این بیماری خواهید داشت یا خیر، کمک کند (البته اطمینان این غربالگری 100 درصد نیست). نتایج غربالگری اطلاعاتی درباره شدت بروز بیماری در فرزندانتان به شما نمیدهد.
همسر شما نیز باید از نظر احتمال ناقل بودن آزمایش بدهد. اگر همسرتان هم ناقل باشد، احتمال به دنیا آمدن کودک مبتلا به SMA 25 درصد است.
غربالگری قبل از باردار شدن و حین بارداری قابل انجام است. بدیهی است که انجام این آزمایش قبل از بارداری آگاهی بیشتری برای تصمیمگیری به شما میدهد.
اگر قبل از بارداری متوجه شدید که خود و همسرتان ناقل هستید، یک مشاوره ژنتیک میتواند راهنمای خوبی برای انجام برخی اقدامات جهت بارداری باشد. این اقدامات میتواند برنامهریزی برای استفاده از لقاح آزمایشگاهی (IVF) و یا تلقیح داخل رحمی (IUI) باشد. اگر باردار هستید و در اطرافیانتان بیمار مبتلا به SMA دارید، با در نظرگیری فواید در برابر ریسک، آمنیوسنتز یا نمونهبرداری از پرزهای جفت (CVS) میتواند در تشخیص ابتلای فرزندتان کمک کننده باشد. در نهایت ممکن است تصمیم بگیرید که باردار نشوید و یا کودکی را به فرزندخواندگی قبول کنید.
اگر متوجه شدید که ناقل SMA هستید، از آنجا که ممکن است سایر اعضای خانواده نیز در معرض خطر ناقل بودن باشند، متخصص زنان، مشاور ژنتیک یا سایر متخصصان مراقبتهای بهداشتی میتوانند در مورد انتخاب بهترین راه برای اطلاع به نزدیکان به شما کمک کنند.
درمان کاملی برای این بیماری وجود ندارد و فقط میتوان سرعت پیشرفت آن را کند کرده و کیفیت زندگی را بهبود بخشید. انتخاب روش درمانی به نوع SMA، شدت بیماری و در دسترس بودن دارو وابسته است. در حال حاضر، چندین دارو برای بهبود بیماری SMA وجود دارد. این داروها منجر به افزایش میزان پروتئین SMN در بدن می شوند. ریسدیپلام یکی از این داروهاست که در مطالعات بهبود عملکرد حرکتی را نشان داده است.
بسیاری از افراد مبتلا به SMA نیاز به مراقبت شبانهروزی از سوی خانواده خود و متخصصان پزشکی دارند. با توجه به این که مشکلات تنفسی ممکن است برای بیماران رخ دهد، بهتر است بیماران یا مراقبان تکنیکهایی را بیاموزند و تمرین کنند که به حفظ راه هوایی و به حداقل رساندن تاثیر عفونتهای تنفسی کمک کند. از طرف دیگر کاردرمانی میتواند با بهبود تون عضلانی تنه، بازو، پا و گردن به کاهش یا جلوگیری از عوارض پیشرونده بیماری کمک کند. بهرهگیری از دانش متخصص تغذیه و روانشناس نیز میتواند در بهبود سبک زندگی روزانه موثر باشد.